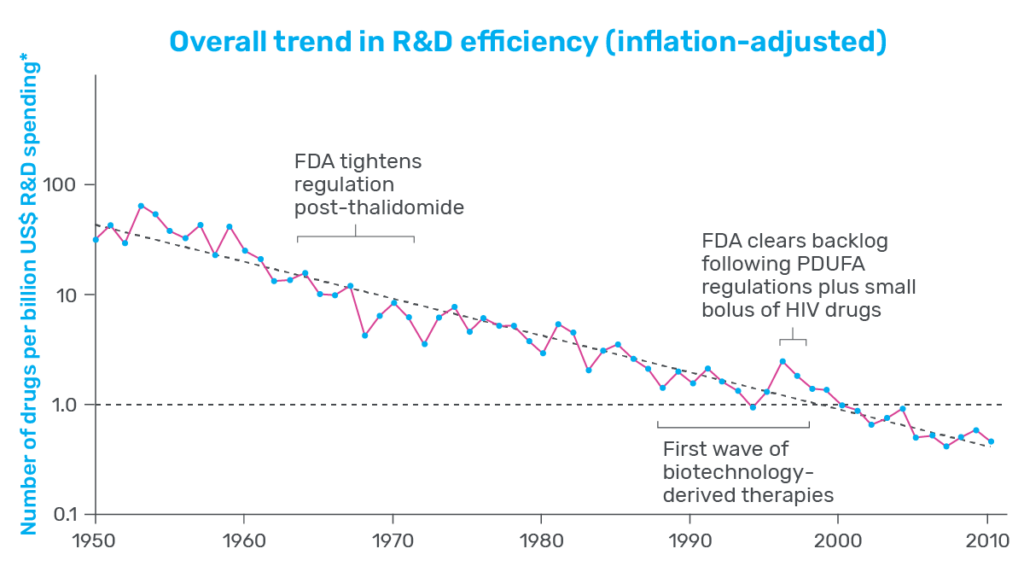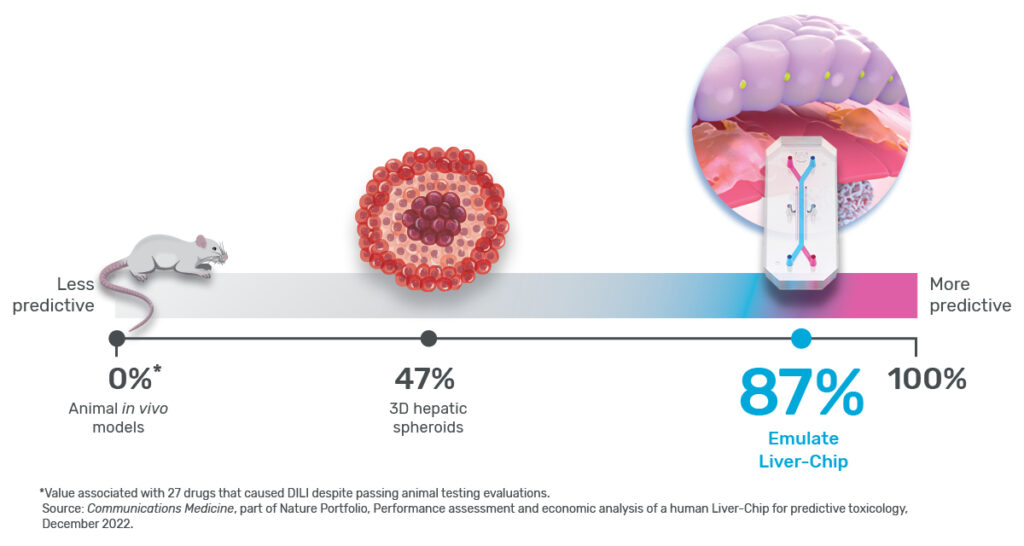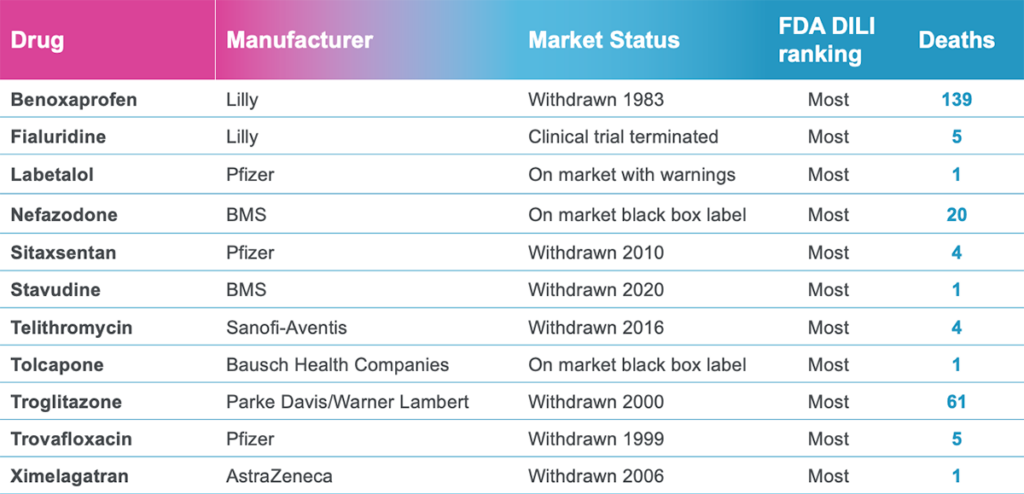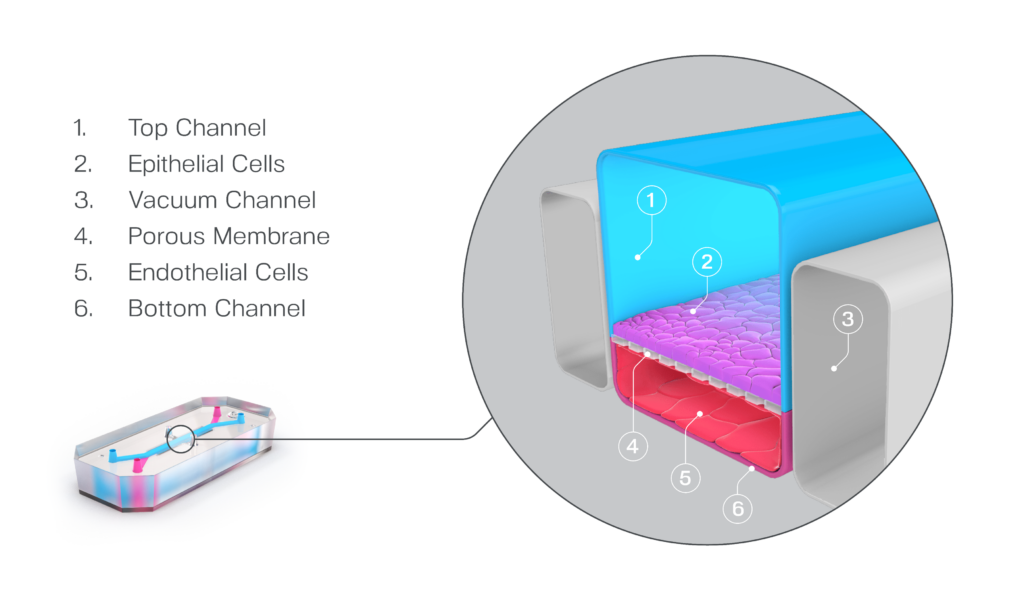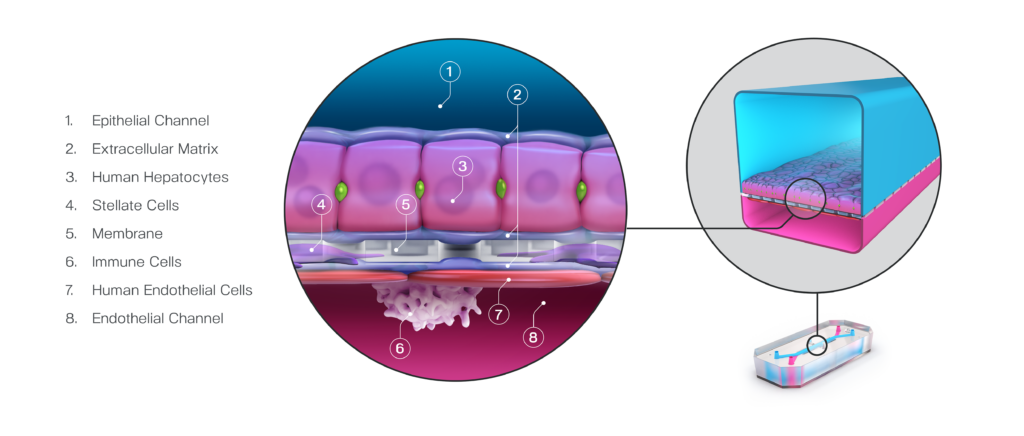
In 2025, momentum around New Approach Methodologies (NAMs) shifted decisively from advocacy to action. Regulatory agencies across the globe took concrete steps to reduce reliance on animal testing and actively encourage the use of more human-relevant technology in drug development. In the U.S., the FDA announced plans to phase out animal testing requirements, starting with monoclonal antibodies and expanding from there, while in the UK and EU, similar policy signals reinforced the need for more predictive, human-based models.
At the same time, it became increasingly clear that the next inflection point for NAMs would not be regulatory acceptance—but practical adoption. Regulatory agencies have already endorsed the use of NAMs and, in some cases, explicitly pointed to Organ-on-a-Chip data as evidence that human-based systems can outperform animal models in predicting clinical response. The remaining challenge now sits with industry: how to integrate these technologies into existing drug development workflows with confidence.
For pharmaceutical teams, the questions are no longer whether Organ-Chips are valid, but where they can be used first to reduce animal studies, how their data can inform real go/no-go decisions, and what level of evidence is needed to trust NAM-derived results alongside—or in place of—traditional animal models. In this context, 2025 produced a remarkable body of Organ-on-a-Chip research that directly addresses these questions—not by promoting novelty, but by demonstrating utility.
Below is a summary of ten of the most impactful Organ-Chip publications from 2025, which we’ve grouped into two complementary categories: 1) how Organ-Chips enable discovery of human-relevant biology that cannot be accessed with traditional models, revealing mechanisms, interactions, and disease signatures that would otherwise remain hidden, and 2) publications that build confidence in practical adoption by demonstrating how Organ-Chips can be benchmarked against in vivo data, clinical outcomes, and established development workflows—providing concrete entry points for reducing animal use while improving decision-making.
Organ-Chip Studies That Uncovered Unique Human-Relevant Insights Not Accessible with Other Models
Reconstructing the Osteolytic Bone Metastatic Niche to Reveal Human-Relevant Drivers of Breast Cancer Progression
Publication: Multi-omics qualification of an organ-on-a-chip model of osteolytic bone metastasis
Summary: This work used a multi-cellular bone metastasis Organ-Chip to model osteolytic breast cancer progression, integrating osteocytes, osteoclasts, and breast cancer cells in a spatially organized, microfluidic system. The study revealed that only the complete tri-culture configuration triggered synergistic activation of inflammatory and pro-metastatic signaling pathways associated with bone destruction. Importantly, transcriptomic signatures from the chip closely aligned with in vivo metastasis datasets, validating biological relevance while preserving experimental control. The impact lies in demonstrating how Organ-Chips can disentangle cell–cell interactions within metastatic niches and identify pathways that may be missed or obscured in animal models.
Why this matters: This study shows how Organ-Chips can recreate complex metastatic microenvironments and uncover invasion and bone-degradation pathways that are difficult to resolve in animal models or simplified in vitro systems.
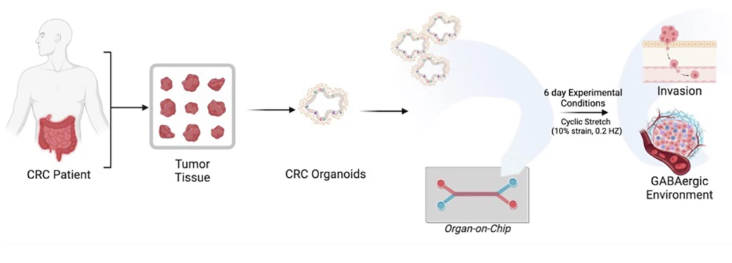
Linking Clinical Outcomes to Tumor Invasion Mechanisms Using Patient-Derived Colorectal Cancer Organ-Chips
Publication: GABAergic signaling contributes to tumor cell invasion and poor overall survival in colorectal cancer
Summary: Starting from clinical trial datasets, this study identified GABAergic signaling as a marker of poor prognosis in metastatic colorectal cancer, then used patient-derived tumor Organ-Chips to interrogate the underlying biology. The chip model demonstrated that tumor-derived GABA directly promotes invasion and that inhibiting GABA synthesis reduced invasive behavior. Notably, organoid-on-chip systems captured patient-specific heterogeneity more faithfully than organoids alone. The impact of this work lies in showing how Organ-Chips can function as mechanistic extensions of clinical data—connecting correlation to causation in a human-relevant context.
Why this matters: By connecting patient survival data to mechanistic invasion pathways on-chip, this work demonstrates how Organ-Chips can bridge clinical observation and causality in human cancer biology.
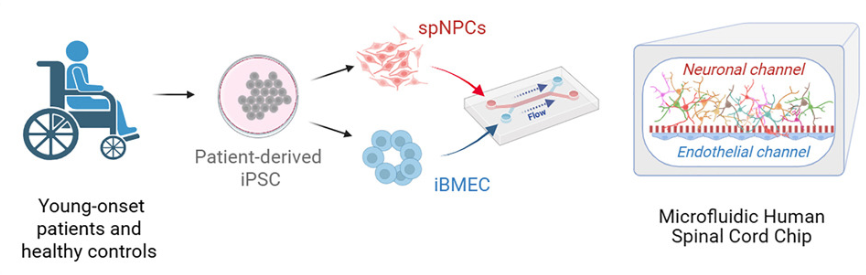
Modeling Early Sporadic ALS Pathology in a Human Spinal Cord Organ-Chip with an Integrated Vascular Interface
Publication: An organ-chip model of sporadic ALS using iPSC-derived spinal cord motor neurons and an integrated blood-brain-like barrier
Summary: Using patient-derived iPSCs, this spinal cord Organ-Chip recreated key aspects of the motor neuron microenvironment, including a functional vascular interface. Multi-omics analysis uncovered early ALS-associated molecular changes—such as neurofilament dysregulation and synaptic signaling defects—before overt neuron loss occurred. These findings mirror clinical biomarkers observed in patients but are difficult to detect in animal models. The impact is significant: the study shows how Organ-Chips can reveal early disease mechanisms in complex, sporadic conditions and provide a platform for earlier, more targeted therapeutic intervention.
Why this matters: The study reveals early, human-specific molecular and functional disease signatures in sporadic ALS that are inaccessible in traditional neuronal cultures or animal models.
Revealing Protective Cervicovaginal Crosstalk by Linking Human Cervix and Vagina Organ-Chips
Publication: Cervical mucus in linked human Cervix and Vagina Chips modulates vaginal dysbiosis
By linking Cervix and Vagina Organ-Chips, this study showed that cervical mucus plays an active protective role in modulating vaginal inflammation and epithelial injury during dysbiosis. Exposure to cervix-derived mucus reduced inflammatory responses and altered protein expression profiles in the vaginal epithelium, identifying potential biomarkers and therapeutic targets for bacterial vaginosis. The broader impact lies in proving that Organ-Chips can model dynamic, multi-organ interactions—unlocking physiological insights that static or single-organ systems cannot provide.
Why this matters: This work demonstrates how multi-organ Organ-Chip systems can capture inter-tissue communication and uncover protective physiological mechanisms that cannot be studied in isolated organ models.
Organ-Chip Studies That Build Confidence in Translational and Regulatory Relevance
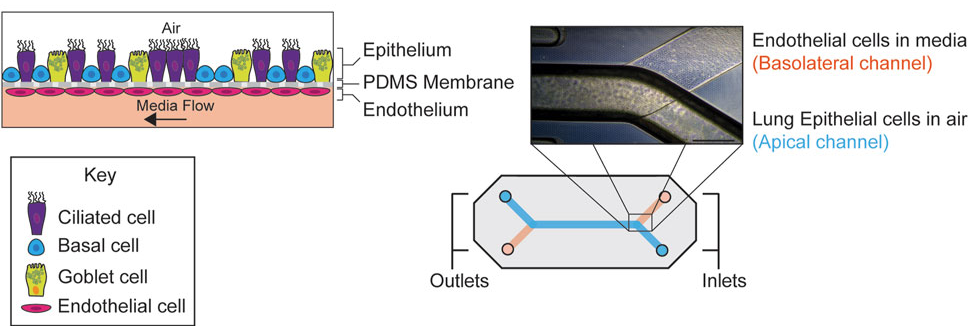
Quantitative Evaluation of Drug Permeability Using a Human Airway Lung-Chip in a Regulatory Research Setting
Publication: Assessment of drug permeability using a small airway microphysiological system
Summary: Conducted within an FDA research laboratory, this study evaluated the Airway Lung-Chip for measuring permeability of inhaled drugs across a differentiated human airway epithelium. The model reproduced key airway structures and generated compound-specific permeability profiles consistent with known clinical behavior. Its significance lies not just in the data, but in the context: it represents regulatory-facing validation of Organ-Chips as credible tools for drug evaluation.
Why this matters: This FDA-led study establishes Organ-Chips as fit-for-purpose tools for generating quantitative, regulator-relevant permeability data in human airway models.
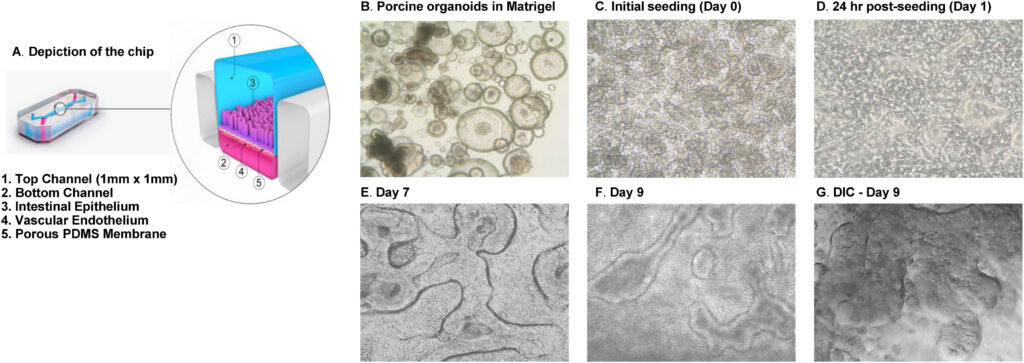
A Porcine Intestine Organ-Chip for Mechanistic Assessment of Drug Transport, Metabolism, and Barrier Function
Publication: Porcine intestinal organoids cultured in an organ-on-a-chip microphysiological system
Summary: This work integrated porcine intestinal organoids into an Organ-Chip platform, producing a highly differentiated epithelium with functional transporters and metabolizing enzymes. The system enabled mechanistic studies of permeability, metabolism, and barrier recovery that are challenging in traditional models. The impact is practical: it demonstrates how Organ-Chips can support non-rodent testing strategies aligned with human physiology and regulatory expectations.
Why this matters: The study shows how Organ-Chips can extend beyond rodent models to support translationally relevant, non-rodent testing in pharmaceutical development workflows.
Dissecting Species-Specific Toxicity Mechanisms Using Parallel Human and Non-Human Organ-Chip Systems
Publication: Rat and dog quad-culture liver chip models: characterization and use to interrogate a potential flavin-containing monooxygenase-mediated, species-specific toxicity of a histamine receptor antagonist
Summary: By directly comparing human and non-human Organ-Chip systems, this study distinguished species-specific toxicities from effects likely to translate to humans. This capability is critical when animal data are ambiguous or conflicting. The impact lies in positioning Organ-Chips not merely as alternatives, but as tools for interpreting and de-risking preclinical safety signals.
Why this matters: This work demonstrates how Organ-Chips can clarify whether preclinical toxicity findings are human-relevant, reducing uncertainty in safety decision-making.
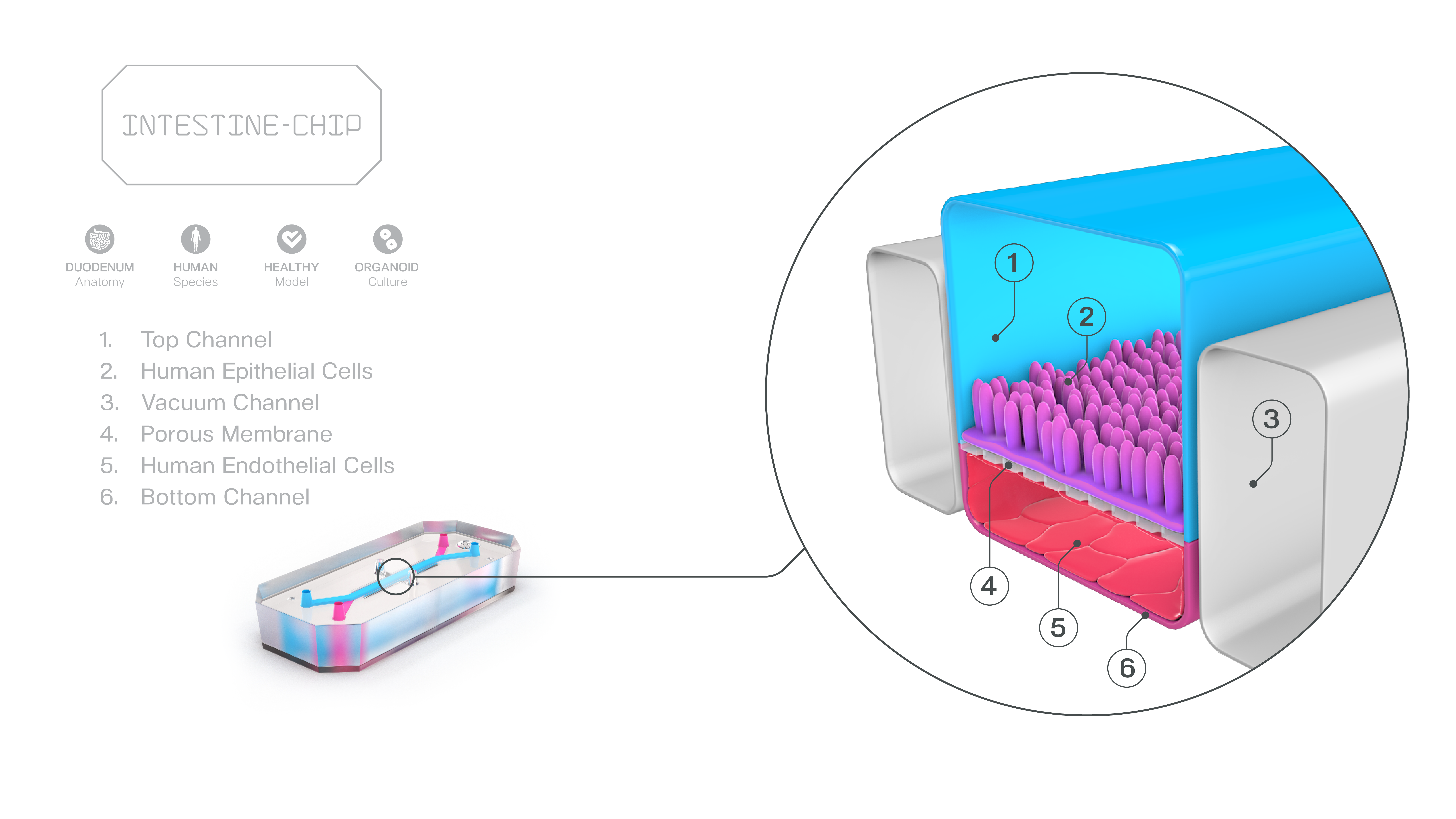
Integrating Human Duodenum Organ-Chip Data with PBPK Modeling to Improve Prediction of Oral Drug Absorption
Publication: Establishing the Human Duodenum Chip as a Surrogate for Effective Human Permeability: In Vitro and In Silico Assessment
Summary: This work demonstrated that permeability values generated in a human Duodenum Organ-Chip strongly correlate with human effective permeability and outperform conventional static models. When integrated into PBPK platforms, the data accurately predicted clinical exposure. The impact is clear: this study shows how Organ-Chips can slot directly into existing computational workflows to support regulatory-aligned, animal-sparing drug development.
Why this matters: By linking Organ-Chip data directly to in silico models, this study provides a practical framework for replacing animal permeability studies with human-relevant NAM workflows.
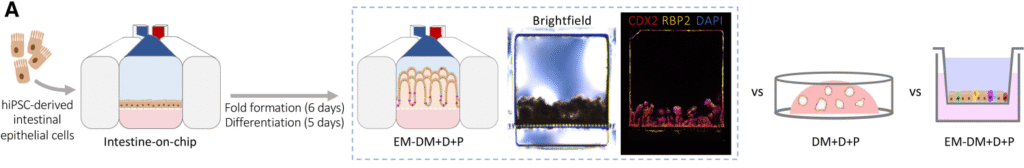
Culture Format Determines Intestinal Maturation: A Head-to-Head Comparison of Organoids, Transwells, and Organ-Chips
Publication: Gene expression profiling reveals enhanced nutrient and drug metabolism and maturation of hiPSC-derived intestine-on-chip relative to organoids and Transwells
Summary: By comparing hiPSC-derived intestinal cells across organoids, Transwells, and Organ-Chips, this study showed that the Organ-Chip format consistently produced the most mature, functionally relevant intestinal phenotype. Genes associated with digestion, nutrient transport, and drug metabolism were most strongly expressed on-chip, while other systems retained more fetal-like characteristics. The impact is foundational: it demonstrates that microenvironment—not just cell source—determines model relevance.
Why this matters: The findings show that Organ-Chips uniquely drive adult-like intestinal function, reinforcing their value as more predictive alternatives to static culture systems.
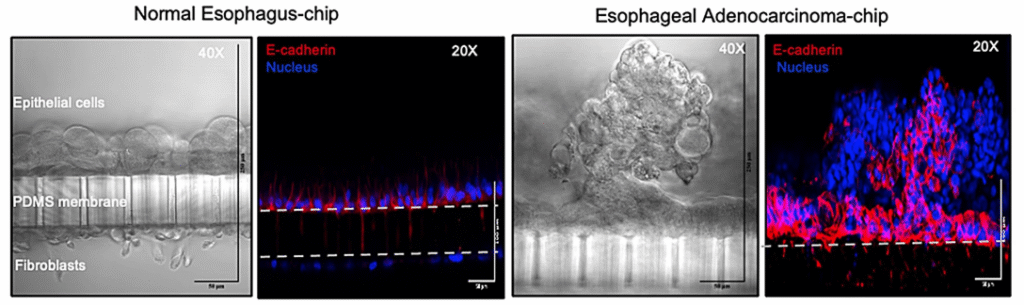
Predicting Patient-Specific Chemotherapy Response Using a Patient-Derived Esophageal Cancer Organ-Chip
Publication: Patient-derived esophageal adenocarcinoma organ chip: a physiologically relevant platform for functional precision oncology
Summary: Using patient-derived tumor material, this Organ-Chip model accurately predicted individual responses to neoadjuvant chemotherapy within weeks of biopsy—outperforming static organoid cultures. The chip preserved tumor–stroma interactions and enabled clinically realistic drug delivery. The impact lies in showing how Organ-Chips can function as patient “avatars,” supporting faster, more informed treatment decisions and translational oncology research.
Why this matters: This study demonstrates how Organ-Chips can deliver clinically actionable predictions on patient timelines, advancing functional precision oncology beyond organoids alone.
Looking Ahead: From Proof to Practice
Taken together, these publications illustrate why 2025 marked a turning point for Organ-on-a-Chip technology. Across discovery, safety, pharmacokinetics, and precision medicine, Organ-Chips demonstrated not only biological relevance, but decision-making value. As regulatory agencies continue to encourage NAM adoption, the evidence base is no longer hypothetical—it is operational, quantitative, and increasingly indispensable.
The question facing industry is no longer whether Organ-Chips can work, but where they can deliver the greatest impact next.