Learn about the importance of sensitivity in drug development and why researchers can improve their pipelines by using more sensitive models.
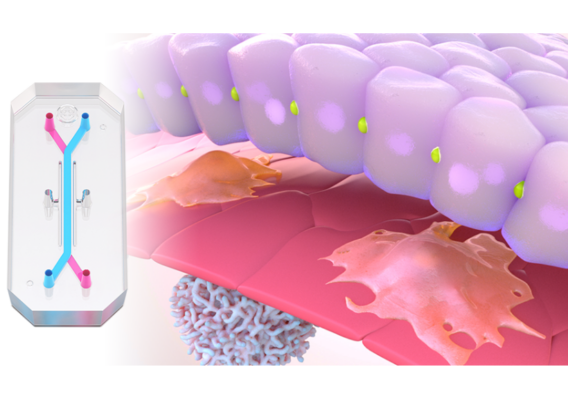
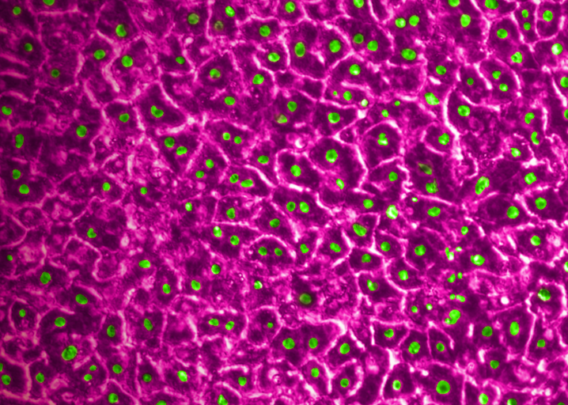
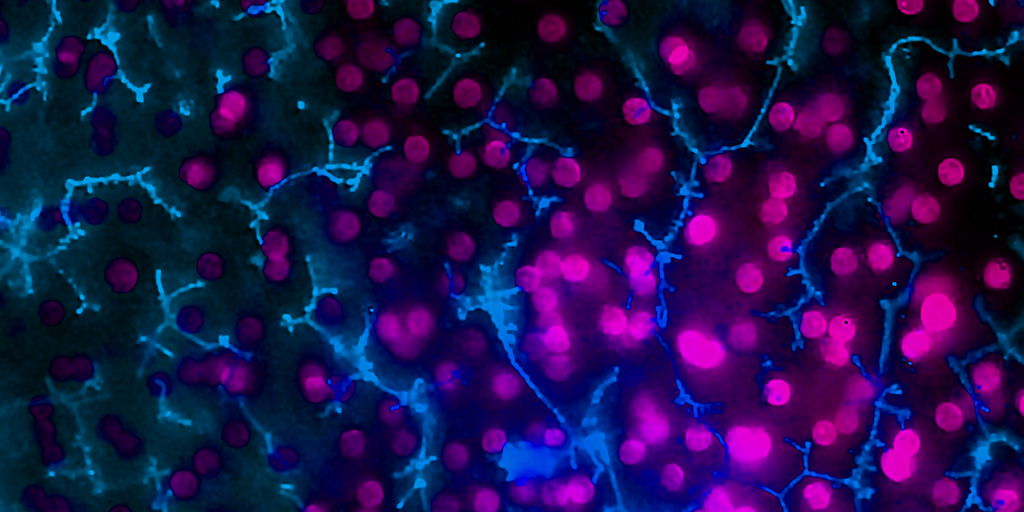
In a recently published study in Communications Medicine, part of Nature Portfolio, Emulate scientists reported that the human Liver-Chip—an advanced, three-dimensional culture system that mimics human liver tissue—could have saved over 240 lives and prevented 10 liver transplants that were caused by the test set of drugs. Specifically, the study demonstrated that the Emulate human Liver-Chip could be used to identify a candidate drug’s likelihood of causing drug-induced liver injury (DILI), a leading cause of safety-related clinical trial failure and market withdrawals around the world1,2.
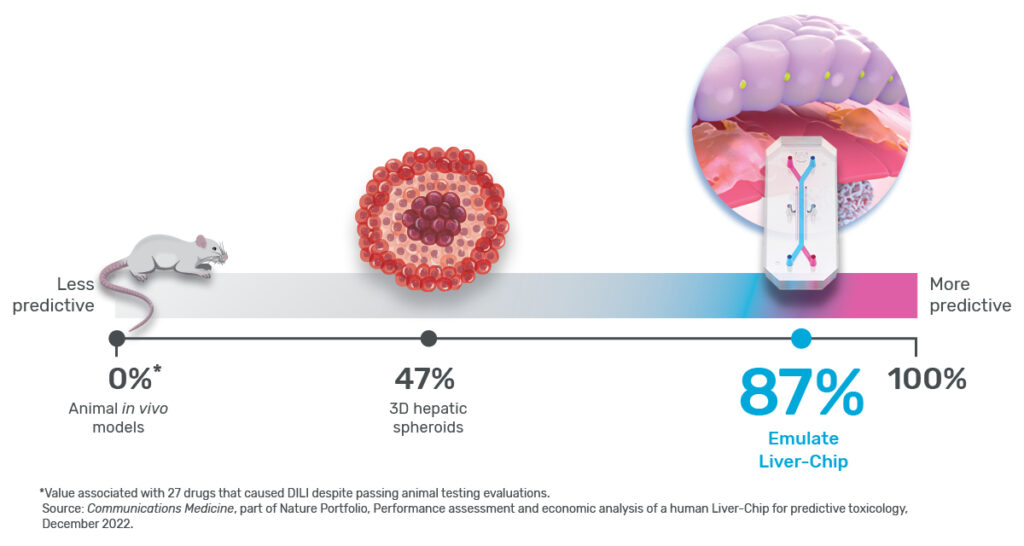
Any preclinical model that helps prevent toxic drugs from reaching patients would be extremely useful, but exactly how much so depends in part on its sensitivity. In the context of predictive toxicology, sensitivity is a measure of how well a model can identify a drug candidate’s toxicity. For example, a model with 0% sensitivity would fail to identify every toxic drug it encounters, whereas one with perfect 100% sensitivity would never miss.
Ewart et al. concluded that the Emulate human Liver-Chip could profoundly affect preclinical drug development because it showed a high sensitivity of 87%—specifically for identifying drugs that were cleared for clinical trials after being tested in both animals and in vitro systems, but ultimately proved hepatotoxic in humans. In other words, the Liver-Chip identified DILI risk for nearly 7 out of every 8 hepatotoxic drugs it encountered.
That’s striking—particularly given the drugs that were being tested: Each had previously made it into the clinic, meaning they were deemed safe enough to administer to humans based on rigorous preclinical testing. While the specifics of preclinical testing may differ for each drug, each includes testing in at least two animal species per regulatory guidelines to progress a drug candidate into clinical testing. Despite this, 22 of the drug candidates went on to be proven toxic in patients. Because animal models failed to adequately forecast the harm that these drugs could—and did—bring to human patients, more than 240 people lost their lives, and 10 were forced to undergo emergency liver transplantation.
Expressed in the language of preclinical toxicology, because the tested drugs made their way through animal testing, animals served as the “reference” in the study, meaning a 0% sensitivity for this drug set—a strong contrast to the human Liver-Chip’s 87%. While it’s important to bear in mind that sensitivity will depend upon the reference set of drugs—as it did here—the juxtaposition of these numbers tells an important story about modern drug development and its ability to protect patients. But to appreciate this story, it’s necessary to scrutinize the concept of model sensitivity and the assumptions underlying it.
The Assumption of Sensitivity
As mentioned above, sensitivity measures how often a model system correctly identifies a drug as toxic or, conversely, how often it incorrectly marks a toxic drug as not toxic. If the model system allows a toxic drug to pass through without a strong indication of harm, it will support a false conclusion that the drug is non-toxic—a result colloquially known as a “false-negative.”
False-negatives can have dire consequences, enabling harmful drug candidates to reach the patient’s bedside through clinical trials. To avoid this, researchers have long sought to test candidate drugs in model systems that closely approximate the human body and prevent bad drugs from reaching the market. And for more than 80 years, animals have filled that need. However, animals are far from perfect. Genetic and physiological differences can produce nuanced yet significant discrepancies in how animals respond to drugs—a drug that appears safe in rats may turn out to be lethal in humans. Such a result would be described as a false-negative.
As roughly 90% of drugs that enter clinical trials fail—many due to safety concerns—it is clear that animal models are far from 100% sensitive3.
So how sensitive are they? The short answer is that, although animals have been tested for the better part of a century, researchers have yet to produce robust data on the sensitivity of animal models, particularly with respect to preclinical toxicology screening4. Perhaps the largest hurdle preventing them from making such an assessment is their assumption that animals are as good as it gets—a belief that eclipses many researchers’ desire for proof.
Because there is a lack of firm data, it’s difficult to make general estimates on animal models sensitivity. However, about 90% of clinical trials fail, and 30–40% of these failures are due to toxicity responses. From this, it can be presumed that animals did not provide sufficient evidence to forecast drug toxicity in humans and that including more sensitive models could have helped prevent toxic drugs from reaching humans.
Stay Up to Date with the Emulate newsletter
The Story in The Numbers
Animal models are meant to be a protective barrier, the last line of defense that prevents toxic drug candidates from reaching humans. But, as described above, this barrier is imperfect—it has gaps in sensitivity that allow toxic drugs to advance into clinical trials far too often.
In their study, Ewart et al. evaluated whether the Emulate human Liver-Chip could fortify this barrier. The team selected 27 drugs, 22 of which were known to be hepatotoxic. Importantly, many of these drugs were included in a set of guidelines that the IQ Consortium, an affiliate of the International Consortium for Innovation and Quality in Pharmaceutical Development, has designated as a baseline researchers should use to measure a liver model’s ability to predict DILI risk.
Each of these drugs had previously advanced to clinical trials, and some were market approved. In other words, animal models in preclinical testing served as the reference for the study and therefore had a 0% sensitivity for this group of drugs.
To measure a model’s sensitivity, researchers use it to screen a set of test drug candidates. To effectively gauge the model’s sensitivity, it’s essential that the set of drugs be carefully selected. For example, one could bias the test drugs towards “easy” drugs—ones that are very toxic in ways that even simple models would identify; however, showing that a new model is excellent at capturing “easy” drugs likely does not demonstrate its utility in capturing the difficult drugs that slip through animal testing. Establishing sensitivity based on such drugs is akin to claiming that a telescope’s ability to spot the sun makes it sensitive to observing stars—it may be technically true but is irrelevant to real-world challenges. That said, it’s not uncommon for preclinical models to be simply tested using highly toxic drugs that never made it to clinical trials5,6.
Stated plainly, a model’s sensitivity changes depending on the drugs that are being tested. If you use clearly toxic drug candidates—ones that would never make it past animal models to begin with—your model will likely appear very sensitive because these drugs are easy to detect. But, if one achieves a high sensitivity when using more challenging drugs—ones that slip through and only show their toxic potential in humans—then the sensitivity of that model will be far more valuable.
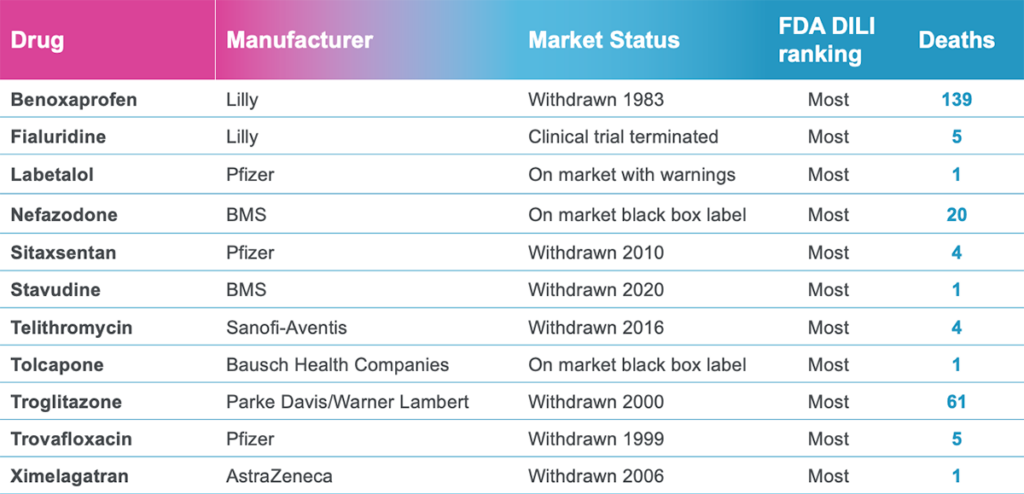
This is precisely why Ewart et al.’s study stands out. Instead of testing Liver-Chips with drugs that are too toxic for animal use, the researchers relied on drugs that had already undergone animal testing and appeared safe. This particular set of drugs not only bypassed animal testing, but they also went on to kill 242 patients. The Liver-Chip successfully identified all of them (see Figure 2). As an additional challenge, they also tested a set of drugs that hepatic spheroid models missed. Even though these drugs were especially difficult for current models to detect, the Liver-Chip achieved an impressive sensitivity of 87%.
By producing these results, Ewart et al. have shown that the human Liver-Chip can, at a minimum, help fill the sensitivity gaps that plague animal models. Stated another way, this data strongly suggests that Liver-Chips can help to identify toxic drugs that animal models miss, which could greatly reduce the number of harmful drugs that advance into clinical trials.
These numbers tell a story about modern and future drug development. Today, drug developers rely on models that, while useful, are imperfect. This imperfection can be catastrophic for patients, as it was for the patients who lost their lives to the drugs Ewart et al. tested. But, in recognizing these imperfections, we can work to prevent future harm by building a better drug development process—one fortified by modern technologies like the Liver-Chip.
References
- Craveiro, Nuno Sales, et al. “Drug Withdrawal due to Safety: A Review of the Data Supporting Withdrawal Decision.” Current Drug Safety, vol. 15, no. 1, 3 Feb. 2020, pp. 4–12, https://doi.org/10.2174/1574886314666191004092520.
- Research, Center for Drug Evaluation and. “Drug-Induced Liver Injury: Premarketing Clinical Evaluation.” U.S. Food and Drug Administration, 17 Oct. 2019, www.fda.gov/regulatory-information/search-fda-guidance-documents/drug-induced-liver-injury-premarketing-clinical-evaluation.
- Fogel, David B. “Factors Associated with Clinical Trials That Fail and Opportunities for Improving the Likelihood of Success: A Review.” Contemporary Clinical Trials Communications, vol. 11, Sept. 2018, pp. 156–164, www.ncbi.nlm.nih.gov/pmc/articles/PMC6092479/, https://doi.org/10.1016/j.conctc.2018.08.001.
- Bailey, Jarrod, et al. “An Analysis of the Use of Animal Models in Predicting Human Toxicology and Drug Safety.” Alternatives to Laboratory Animals, vol. 42, no. 3, June 2014, pp. 181–199, https://doi.org/10.1177/026119291404200306.
- Zhou, Yitian, et al. “Comprehensive Evaluation of Organotypic and Microphysiological Liver Models for Prediction of Drug-Induced Liver Injury.” Frontiers in Pharmacology, vol. 10, 24 Sept. 2019, https://doi.org/10.3389/fphar.2019.01093. Accessed 22 Nov. 2020.
- Bircsak, Kristin M., et al. “A 3D Microfluidic Liver Model for High Throughput Compound Toxicity Screening in the OrganoPlate®.” Toxicology, vol. 450, Feb. 2021, p. 152667, https://doi.org/10.1016/j.tox.2020.152667.